диссертация (1105558), страница 19
Текст из файла (страница 19)

гидразон
Рис. 10. Хроматограмма 1,1-диметилгидразона п-НБА. Концентрация гидразона 0,01 мг/л. Элюент 50 мМ раствор ацетата аммония и 60% ацетонитрила. Детектор амперометрический.
Зависимость площади пика гидразона от концентрации НДМГ линейна в диапазоне концентраций 0,05-2,5 мг/л. Рассчитаны пределы обнаружения 1,1-диметилгидразона
4-нитробензальдегида с использованием спектрофотометрического (СФ) и амперометрического (АД) детекторов, основные метрологические характеристики определения гидразона представлены в табл. 12.
Таблица 12. Основные метрологические характеристики определения гидразона. Вид зависимости у=ах+b
| Параметры | СФ | АД |
| Коэффициент a | 180±8 | 42,4±0,9 |
| Коэффициент b | 7,6±0,4 | 0,22± 0,02 |
| r | 0,9998 | 0,9999 |
| Диапазон линейности, мг/л | 0,1-2,5 | 0,05-2,5 |
| sr (n=3, P=0,95) | 0,05 | 0,05 |
| Cmin, мг/л | 0,045 | 0,005 |
4.1.3. Сорбционное концентрирование гидразона п-НБА
Для уменьшения предела обнаружения НДМГ использовали сорбционное концентрирование в off-line режиме на картриджах Strata C18-E (Phenomenex). Контроль сорбции показал количественное удерживание гидразона в фазе сорбента при составе пробы, соответствующем условиям дериватизации. Установлено, что для количественной десорбции гидразона необходимо 3 мл ацетона. В случае гидразона п-НБА добиться совместимости элюента, используемого для десорбции концентрата, с подвижной фазой не удается, так как добиться количественной десорбции гидразона с картриджа приемлемым объемом подвижной фазы не представляется возможным. Поскольку макросостав концентрата после десорбции должен быть идентичен составу подвижной фазы для хроматографического разделения, необходимо проводить смену растворителя, которую осуществляли путем отгонки ацетона на роторном испарителе и перерастворения остатка в 1 мл подвижной фазы.
Для изучения эффективности концентрирования в картридж доставляли различные объемы модельного раствора с постоянным содержанием определяемого компонента - 10 мкг/л. Далее проводили элюирование вещества из фазы сорбента и его растворение с использованием постоянных объемов соответствующих растворителей. На рисунке 11 приведена зависимость площади хроматографического пика от степени концентрирования.

Рис. 11. График зависимости площади пика гидразона от коэффициента концентрирования. Элюент 50 мМ ацетат аммония и 60% ацетонитрила. Детектор амперометрический. Уравнение прямой: y=2,245x-12,682, r=0,997.
Из графика видно, что интенсивность аналитического сигнала линейно возрастает при увеличении степени концентрирования в диапазоне k 5-120. Это позволяет осуществлять концентрирование ультрамалых количеств гидразона в диапазоне 0,05-5 мкг/л с объемом пробы 100мл.
Сорбционно-хроматографическое определение НДМГ проводили в диапазоне 0,05-2,0 мкг/л. Объем пробы при концентрировании составил 100 мл, а после десорбции производного и его перерастворении – 1 мл. Установлено, что при концентрациях 0,05-0,5 мкг/л НДМГ не наблюдается линейной зависимости площади пика от концентрации. По-видимому, при ультрамалых концентрациях реакция дериватизации протекает с низким выходом из-за обратимости реакции образования гидразонов, а также снижения скорости протекания реакции при малых концентрациях НДМГ. К тому же, большое количество побочных продуктов реакции затрудняют определение гидразона при его содержании менее 0,5 мкг/л. Хроматограмма пробы, содержащей 0,5 мкг/л НДМГ после реакционного сорбционно-хроматографического определения с наиболее чувствительным амперометрическим детектированием представлена на рис. 12.

гидразон
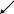
Рис. 12. Хроматограмма 1,1-диметилгидразона п-НБА, полученная при реакционном сорбционно-хроматографическом определении НДМГ. Концентрация НДМГ 0,5 мкг/л. Элюент 50 мМ раствор ацетата аммония и 60% ацетонитрила. Детектор амперометрический.
Таким образом, сорбционно-хроматографическое определение НДМГ в виде гидразона п-НБА не обеспечивает низких пределов обнаружения НДМГ даже при амперометрическом детектировании. А сам подход нельзя автоматизировать из-за необходимости замены матрицы после десорбции концентрата. Поэтому требуется поиск других экспрессных реагентов, обеспечивающих возможность селективного и чувствительного детектирования производных НДМГ, например, флуоресцентных ароматических альдегидов.
4.2. Дериватизация и определение НДМГ с коричным альдегидом
4.2.1. Выбор условий проведения дериватизации
Реакция взаимодействия НДМГ с коричным альдегидом протекает согласно представленной схеме:
 .
.
В ходе реакции образуется диметилгидразон коричного альдегида. Из литературных данных [ ] известно, что образующийся гидразон поглощает при 370 нм, а исходный реагент — при 260 нм. Максимальная оптическая плотность достигается при содержании в реакционной смеси уксусной кислоты и 80% этиленгликоля. Спектрофотометрическое и флуориметрическое определение НДМГ затруднено при анализе реальных объектов в связи с низкой селективностью методов. Для разделения компонентов пробы в варианте ОФ ВЭЖХ необходимо выбрать условия образования диметилгидразона коричного альдегида, совместимые с дальнейшим хроматографическим определением, так как условия, подобранные авторами работы [ ] для определения НДМГ в виде производного с коричным альдегидом методом спектрофотометрии (80% этиленгликоля, ледяная уксусная кислота), не могут использоваться в хроматографической системе. Согласно существующим данным, диметилгидразон коричного альдегида является флуорофором. Использование флуоресценции для детектирования в ВЭЖХ часто бывает предпочтительнее, поскольку она обеспечивает наименьшие пределы обнаружения среди обычно используемых детекторов. Поэтому представляется перспективным разработать способ определения НДМГ методом ВЭЖХ с флуориметрическим детектированием.
Для оценки возможности хроматографического определения НДМГ в виде производного с коричным альдегидом методом ВЭЖХ изучены спектральные характеристики диметилгидразона коричного альдегида. На рис. 13 представлен спектр поглощения диметилгидразона коричного альдегида, снятый в кювете с длиной оптического пути 1 см относительно холостого раствора. Как видно из рисунка, максимум поглощения соответствует 400 нм.
При флуориметрическом детектировании гидразона установлено, что максимум возбуждения флуоресценции диметилгидразона соответствует 328 нм, максимум эмиссии флуоресценции — 398 нм (рис. 14).

Рис. 13. Спектр поглощения производного НДМГ с коричным альдегидом в среде аммонийно-ацетатного буферного раствора (рН 5.4), 50% ацетонитрила. l = 1 см.

Рис. 14. Спектр возбуждения и эмиссии флуоресценции диметилгидразона коричного альдегида. Колонка Nucleosil 5-C18, 4.6 × 150 мм. Подвижная фаза: A — ацетонитрил/Б — 0.05М CH3COOH (pH 3.4), градиентное элюирование: 0–30 мин 45% А, 30–34 мин 90% А, 34–40 мин 45% А. Скорость потока 1 мл/мин.
Так как интенсивность флуоресценции зависит от условий проведения реакции: рН буферного раствора, природы растворителя, его содержания, – то влияние всех этих факторов изучали параллельно с выбором условий максимального выхода реакции.
В связи с тем, что коричный альдегид нерастворим в воде, в ходе работы установлено, что минимальный объем органического растворителя, который должна содержать реакционная смесь для его растворения, составляет 50%. В качестве растворителя использовали ацетонитрил, чтобы приблизить состав реакционной смеси к составу используемой подвижной фазы.
На рис. 15 представлена зависимость аналитического сигнала от содержания ацетонитрила в реакционной смеси. Содержание растворителя варьировали в диапазоне 50-80%, так как 50% ― это минимальное количество, необходимое для растворения реагента, а при содержании ацетонитрила выше 80% сокращается время удерживания диметилгидразона, к тому же происходит сильное разбавление пробы. Согасно рис. 15, 50% ацетонитрила обеспечивают максимальную интенсивность флуоресценции.

Рис. 15. График зависимости площади хроматографического пика диметилгидразона коричного альдегида от содержания ацетонитрила в реакционной смеси.
Влияние природы растворителя изучали на примере использования ацетонитрила, диметилформамида (ДМФА), этанола и изопропанола. Как видно из рис. 16, выбор растворителя не оказывает существенного влияния на интенсивность флуоресценции гидразона. Наилучшие результаты достигаются при использовании ацетонитрила.

Рис. 16. Влияние природы растворителя на площадь хроматографического пика производного НДМГ с коричным альдегидом.
Одним из основных факторов, влияющих на выход продукта реакции конденсации гидразинов с карбонильными соединениями, является рН реакционной смеси. Известно, что график «рН - скорость образования» имеет максимум, связанный с тем, что имеют место два противоположных эффекта: общий кислотный катализ реакции присоединения и уменьшение концентрации свободного азотистого основания при превращении его в сопряженную кислоту [5]. При этом максимум наблюдается при рН, близких к рКа соответствующего гидразина.
Влияние рН на площадь хроматографического пика диметилгидразона в нашей работе изучали на примере использования аммонийно-ацетатных буферных растворов (рН 3.5–6.3) и фосфатного буферного раствора (рН 7.0). Приведенная на рис. 17 зависимость показала, что выход продукта реакции возрастает при увеличении рН до значения 5.4, а затем не зависит от рН. Таким образом, оптимальным значением является рН 5.4, что хорошо согласуется с теоретическими представлениями.

Рис. 17. График зависимости площади хроматографического пика производного НДМГ от рН буферного раствора для проведения реакции. Колонка Nucleosil 5-C18, 4.6 × 150 мм. Элюент: 60% 0.05М Н3PO4/40% ACN. Скорость потока 1 мл/мин. Спектрофотометрический детектор.
Существенное влияние рН реакционной смеси оказывет и на интенсивность флуоресценции, так как при разных рН может происходить стабилизация или дестабилизация электронной структуры соединения, которая и определяет флуоресцентное поведение определяемого вещества. Как видно из рис. 18, максимальная интенсивность флуоресценции наблюдается при рН 5.4, который соответствует оптимальному рН образования диметилгидразона.

Рис. 18. График зависимости площади пика диметилгидразона коричного альдегида от рН буферного раствора для проведения реакции. Флуориметрический детектор.
Варьирование природы буферного раствора на примере использования аммонийно-ацетатного, фосфатного и цитратного буферных растворов с рН 5.4–5.5 не показало выраженной зависимости площади пика диметилгидразона (флуориметрическое детектирование) от природы используемого буферного раствора (табл. 13), поэтому выбран аммонийно-ацетатный буферный раствор как обладающий максимальной буферной емкостью при рН 5.4.
Таблица 13. Влияние природы буферного раствора на площадь хроматографического пика производного НДМГ с коричным альдегидом
| Буферный раствор | S, отн. ед. |
| Аммонийно-ацетатный, рН 5.4 | 21.5 |
| Дигидрофосфатный, рН 5.5 | 21.1 |
| Цитратный, рН 5.4 | 18.9 |
В ходе работы установлено, что изменение концентрации аммонийно-ацетатного буферного раствора в диапазоне 0.01–0.1 М не влияет на площадь пика диметилгидразона (рис. 19). В качестве рабочей была выбрана концентрация 0.05 М.

Рис. 19. График зависимости площади хроматографического пика диметилгидразона от концентрации аммонийно-ацетатного буферного раствора.
Из многочисленных публикаций известна способность гидразонов протонироваться в кислых растворах, поэтому мы изучали поведение гидразона коричного альдегида при различных рН. На рис. 20 представлены спектр поглощения исходного диметилгидразона коричного альдегида и спектры поглощения производного при рН 2.5 и 7.4. Видно, что при подкислении реакционной смеси максимум поглощения сдвигается в коротковолновую область, при этом значительно возрастает интенсивность сигнала. При рН образования гидразона (рН 5.4) и выше гидразон находится в незаряженной форме, максимум поглощения которой соответствует 400 нм. При увеличении рН до 7.0, интенсивность уменьшается при неизменности максимума поглощения.
















