
Эпилептические энцефалопатии раннего детского возраста
Криптогенные и/или симптоматические
Эпилептические энцефалопатии раннего детского возраста
Головной мозг ребенка раннего возраста принципиально отличается по своим функциональным и структурным характеристикам от мозга взрослого человека. Эти отличия в целом обусловлены комплексом характерных черт, объединяющим названием которых является термин «незрелость». Именно незрелость детского мозга ответственна за наличие весьма специфичных по своим проявлениям и почти всегда злокачественных резистентных эпилептических синдромов раннего детского возраста. В свою очередь эпилептические припадки, персистирующие при этих синдромах, в своем воздействии на развивающийся мозг ребенка гораздо более губительны по сравнению с аналогичным воздействием на мозг взрослого больного. Это дает основание применять в характеристике ранних детских эпилептических синдромов понятие «энцефалопатии». В соответствии с последней ревизией Международной классификации эпилепсии и эпилептических синдромов и подгруппу ранних детских энцефалопатии включаются ранняя миоклоническая энцефалопатия, ранняя эпилептическая энцефалопатия, синдром Веста, синдром Леннокса—Гасто (табл. 3.8).
Таблица 3.8. Сравнительная характеристика инфантильных энцефалопатий
| Признак | Ранняя миоклоническая эпилепсия | Синдром Отахары | Синдром Веста | Синдром Леннокса-Гасто |
Рекомендуемые материалыFREE Лекция 17 - Опухоли надпочечников (слайды) (Эндокринология) FREE Лекция 17 - Опухоли легких и плевры (слайды) (Факультетская хирургия) FREE Презентация № 7 по теме Болезни системы кровообращения как медико-социальная проблема (Общественное здоровье и здравоохранение) FREE Лекция 07 - Острый панкреатит (слайды) (Факультетская хирургия) FREE ЛЕКЦИЯ №7 (packed) (Хирургическая стоматология) FREE 4661beab2834890981294b504c7ca42d (Оториноларингология) Возраст дебюта | первые 3 мес | первые месяцы | 5—7 мес | 3—5 лет |
| Ложный тип припадков | +/- | +/- | - | + |
| Тонические спазмы | редко | +/- | + | - |
| Циркадность | диффузная | диффузная | после просыпания | диффузная |
| Откликаемость на АКТГ | неубедительная | отсутствует или неубедительная | хорошая | неубедительная |
| Межприступная ЭЭГ | супрессивно- взрывная | супрессивно-взрывная | гипсаритмия | диффузная медленная спайк-волна |
| ЭЭГ-сна | супрессивно- взрывная | - | - | быстрый ритм |
| Приступная ЭЭГ | десинхронизация | десинхрониза-ция | десинхрониза-ция | быстрая синхронизация |
Обозначения: + наличие признака; — отсутствие признака.
Характеризуя ранние возрастзависимые эпилептические энцефалопатии, можно выделить несколько общих особенностей:
• жесткая зависимость от возраста; невозможность возникновения конкретной эпилептической энцефалопатий вне определенного возрастного диапазона;
• наличие патогномоничных и достаточно специфичных по своим кинематическим проявлениям частых «малых» генерализованных эпилептических припадков;
• тяжелые и продолженные во времени эпилептические изменения на ЭЭГ;
• выраженная гетерогенность этиологии;
• частая ассоциация с моторными и ментальными нарушениями;
• весьма значительные затруднения в лечении и относительно неблагоприятный прогноз.
Такое значительное сходство указанных синдромов, а также использование нейроонтогенетического подхода при анализе ранних детских эпилепсий позволяют предположить, что именно ранний возраст, т.е. структурная и функциональная незрелость головного мозга, является основным фактором, детерминирующим развитие заболевания. Сами ранние эпилептические синдромы целесообразно рассматривать как неспецифическую реакцию незрелого мозга на экзо- или эндогенный стресс-фактор (возможно, один и тот же), реализующуюся эпилептическими припадками, тип которых зависит не от специфики фактора, а от степени структурно-функциональной состоятельности мозга.
Ранняя миоклоническая энцефалопатия. Ранняя миоклоническая энцефалопатия (РМЭ) — редкий возрастзависимый эпилептический синдром, впервые описанный J. Aicardi в 1978 г. В большинстве случаев заболевание начинается в возрасте, не превышающем 3 мес. Основным типом припадков являются миоклонии, преимущественно в виде фрагментарного мио-клонуса. Кроме того, могут наблюдаться частые внезапные парциальные приступы, массивные миоклонии и тонические спазмы.
Среди перечисленных клинических проявлений РМЭ патогномоничным признаком следует считать частые фрагментарные миоклонии, которые являются не только самым частым типом приступов, но и считаются дебютным, ранним симптомом заболевания. Тем не менее, с течением заболевания фрагментарные миоклонии постепенно уступают свою ведущую клиническую роль частым парциальным припадкам. Характеризуя особенности миоклонии, можно отметить, что они проявляются не только в состоянии бодрствования, но и во время сна. По степени выраженности они могут варьировать от легкого подергивания дистальных фаланг пальцев рук до миоклонии кистей, предплечий, век и угла рта. Частота их возникновения — от нескольких раз в день до нескольких десятков в минуту.
Парциальные припадки отмечаются приблизительно в 70—80 % случаев РМЭ и, подобно фрагментарным миоклониям, наблюдаются и во время бодрствования, и во время сна. Парциальные приступы при РМЭ отличаются большой частотой и могут достигать 300—500 раз в сутки.
Изменения на ЭЭГ при РМЭ включают специфичный для незрелого детского мозга «супрессивно-взрывной паттерн», состоящий из вспышек продолжительностью 1—5 с, перемежающихся с периодами резкого, почти полного уплощения фоновой активности, длящимися 3—10 с (рис. 3.8). Наиболее ярко супрессивно-взрывной паттерн проявляется в период глубокого сна; S. Ohtahara (1997) напрямую увязывает интенсивность этого выраженного нарушения на ЭЭГ с глубиной сна.
К возрастному периоду 3—5 мес. супрессивно-взрывной ЭЭГ-паттерн постепенно замещается атипичной, или модифицированной, гипсаритмией, хотя в некоторых случаях он может персистировать достаточно долго.
Нейрорадиологические изменения при РМЭ не выражены. Как правило, и КТ, и МРТ не выявляют каких-либо грубых структурных изменений головного мозга. В тех редких случаях, когда они отмечаются, это преимущественно кортикальная атрофия различной степени выраженности.
Относительно этиологических аспектов РМЭ можно отметить, что не выявлено каких бы то ни было специфических этиологических факторов в развитии этого заболевания. Считается [Bernardina D., 1983], что определенную роль могут играть врожденные нарушения метаболизма (дизметабо-лические энцефалопатии раннего возраста), из которых выделяют как особо частые некетотическую гиперглицинемию, пропионовую ацидурию и D-глициновую ацидемию. В основном же большинство случаев РМЭ расценивается как криптогенные, т.е. формы, при которых презумптивно существующая причина, лежащая в основе развития эпилепсии, подразумевается, но не поддается идентификации на современном технологическом Уровне диагностических методов.
Лечение РМЭ составляет тяжелую и пока нерешенную проблему. К сожалению, к настоящему времени не существует антиконвульсантов и гормональных средств, которые могли бы обеспечить сколько-нибудь приемлемую эффективность лечения. Наиболее характерный исход заболевания смерть больных в первые 5 лет жизни; оставшиеся в живых страдают тяжелыми психомоторными расстройствами.
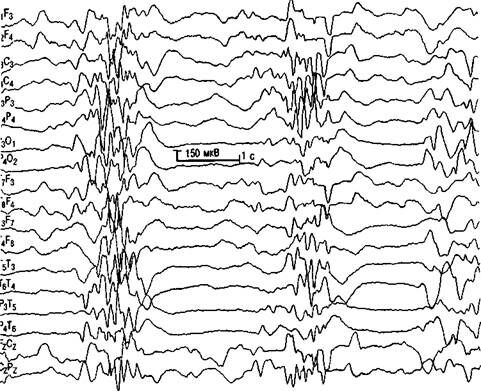
Рис. 3.8. ЭЭГ при РМЭ. Паттерн «вспышка—угнетение».
Ранняя эпилептическая энцефалопатия (синдром Отахары). Эта форма нцефалопатии является самым ранним по дебюту возрастзависимым эпилептическим синдромом. Она была впервые описана в 1978 г. японским ученым Shunsuke Ohtahara, а с 1989 г. признана в качестве самостоятельного эпилептического синдрома, получившего имя своего первооткрывателя — синдром Отахары.
В клинической картине приступы дебютируют в первые или 3 мес. жизни, но особенно часто в 1-й месяц. Основным типом припадков являются серийные или изолированные тонические спазмы. Приступы персистируют не только в состоянии бодрствования, но и ночью, помимо тонических спазмов, почти в половине случаев могут отмечаться моторные парциальные приступы, иногда по гемитипу. Миоклонические припадки нехарактерны, хотя в отдельных случаях могут иметь место.
Продолжительность тонического спазма при синдроме Отахары достигает приблизительно 10 с; в одной серии может отмечаться от 10 до 40 спазмов. Общее суточное количество спазмов достаточно велико и может достигать 300—400.
Основное ЭЭГ-проявление синдрома Отахары — описанный выше супрессивно-взрывной паттерн, который почти всегда отмечается и во сне, и в состоянии бодрствования. Вспышки медленных волн, длящиеся 1—3 с, имеют амплитуду 150—350 мкВ, перемежаются периодами почти полного уплощения ритма продолжительностью 3—4 с (рис. 3.9).
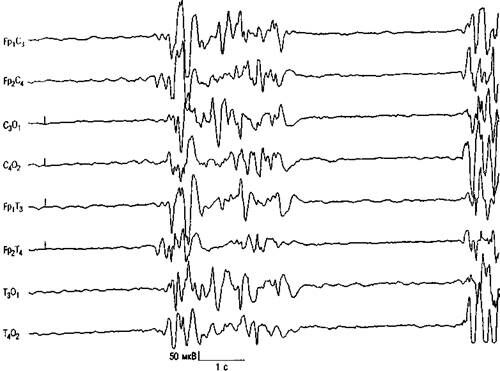
Рис. З.9. Ранняя инфантильная эпилептическая энцефалопатия (синдром Отахары). Диффузная супрессивно-взрывная активность.
В диагностике синдрома Отахары принципиально важны методы нейровизуализации, так как в отличие от РМЭ часто выявляют грубые структурные (порой асимметричные) изменения мозга. По данным S. Ohtahara (1997), эти нарушения отмечаются приблизительно в 85 % случаев ранней эпилептической энцефалопатии.
Как и РМЭ, синдром Отахары полиэтиологичен. В инициации заболевания особую роль играют врожденные мальформации головного мозга.
Синдром Веста. Синдром Веста — возрастзависимый эпилептический синдром, характеризующийся особым типом эпилептических припадков инфантильные спазмы), специфическим вариантом изменений на ЭЭГ гипсаритмия) и задержкой психомоторного развития. Следует отметить, но нозологические определения синдрома Веста достаточно многообразны с течением времени постоянно модифицируются. Одним из последних усовершенствований в терминологии заболевания являются предложения .. Riikonen о принципиальной возможности постановки диагноза синдрома Веста без наличия одного из перечисленных выше признаков. Инфантильные спазмы — это тип эпилептических приступов, представляющих собой массивные миоклонические и(или) тонические, про- и(или) ретропульсивные, симметричные и(или) асимметричные, серийные и(или) изолированные спазмы аксиальной и конечностной мускулатуры. Помимо синдрома Веста, они могут наблюдаться и при других возрастзависимых эпилептических энцефалопатиях: ранней миоклонической энцефалопатии, синдроме Отахары и синдроме Леннокса—Гасто. таким образом, термин «инфантильные спазмы» не отображает нозологи-ескую принадлежность заболевания, свидетельствуя лишь о доминирующи характере припадков.
Мальформативные изменения мозга лежат в основе приблизительно 30—34 % случаев синдрома Веста [Jellinger К., 1987], по данным патоморфологических исследований. По своей классификационной сути это могут быть практически любые пороки развития мозга, включающие агенетические аномалии — агенезию мозолистого тела, агенетические порэнцефалические кисты, голопрозэнцефалию, агенезию червя и(или) гемисфер мозжечка; обширные эмбриоклас-тические процессы — гидранэнцефалию, эмбриокластические порэнцефалические кисты; гипо- и гиперпластические процессы — гипоплазию отдельных долей мозга, микроцефалию, унилатеральную мегалэнцефалию; кортикальные дисплазии — лиссэнцефалию, полимикрогирию, пахигирию, фокальную корковую дисплазию, ленточные и узловые нейронные гетеро-топии, микродисгенезии.
Особая роль в генезе инфантильных спазмов при синдроме Веста в последнее время уделяется микродисгенезиям, которые могут быть легко пропущены при рутинном диагностическом комплексе заболевания [Palm, 1986]. Предполагается, что небольшие участки дисплазированной ткани, Даже в небольшом количестве рассеянные в толще мозгового вещества, способны не только генерировать тяжелые и частые инфантильные спазмы, но и ответственны за грубые ментальные нарушения при синдроме Веста.
Ведущую роль в генезе инфантильных спазмов при этом заболевании безоговорочно отдают пери- и постнатальным гипоксически-ишемическим изменениям мозга [Chevrie, Aicardi, 1977].
Нейропатологические формы гипоксически-ишемических поражений мозга многообразны и, по мнению J.Volpe (1986), объединяются термином шоксически-ишемические энцефалопатии», которые включают селектвный нейрональный некроз; status marmoratus подкорковых ядер, парасагитальный церебральный некроз; перивентрикулярную лейкомаляцию; жальный или мультифокальный ишемически-аноксический церебральный некроз (который в свою очередь включает порэнцефалию и энцефаломаляцию).
Частота инфекционного фактора в развитии заболевания варьирует, по данным различных авторов, от 3 % [Matsumoto et , 1981] до И % [Lombroso C.T., 1983].
Инфекционные заболевания (цитомегалия, токсоплазмоз, герпетичес-й и краснушный энцефалиты, энтеровирусные и аденовирусные энцефалиты) могут играть более значимую роль в генезе инфантильных спазмов, так как вирусологические методы в настоящее время не позволяют проводить объективную оценку. Следует помнить, что свое катастрофическое эпилептогенное влияние они могут оказывать не напрямую, а опосредованно, например, через инициацию различных пороков развития.
Среди других (менее частых) причин возникновения синдрома Веста возмжно упомянуть нарушения метаболизма — до 10 % случаев [Meencke, srhard, 1985]; травматические изменения мозга (родовая травма); различие типы церебральных опухолей [Miyake et al., 1986].
Течение синдрома Веста в зависимости от этиологического фактора. В зависимости от этиологического фактора, лежащего в основе развития инфантильных спазмов при синдроме Веста, можно выделить несколько наиболее типичных типов течения заболевания:
• дальнейшее персистирование инфантильных спазмов в течение нескольких лет. Этот вариант течения заболевания чаще встречается при глубоких нарушениях кортикальной организации (особенно при диффузной лиссэнцефалии) или обширных эмбриокластических процессах (гидранэнцефалии);
• трансформация спазмов в мультифокальную эпилепсию. Как правило, такая процессуальность спазмов отмечается при множественных эпилептогенных нарушениях коры, например при туберозном склерозе или мультифокальной гипоксически-ишемической энцефалопатии. Результатом является появление мультифокальных или вторично-генерализованных приступов, относительно резистентных к антиком вульсантной терапии;
• трансформация спазмов в парциальную эпилепсию. Основные причины подобной трансформации включают порэнцефалию, очаговые формы кортикальных дисплазий и некоторые случаи туберозного склероза с единственным кортикальным туберсом;
• эволюция синдрома Веста в синдром Леннокса—Гасто. Это в основном отмечается при криптогенных инфантильных спазмах, т.е. спазмах, при которых этиологический фактор, лежащий в их основе, не идентифицируется, но подразумевается. Вообще отсутствие явных морфологических изменений мозга при столь губительной для прогноза трансформации инфантильных спазмов в синдром Леннокса— Гасто является одним из удивительных фактов эпилептологии, не поддающихся сколько-нибудь вразумительным объяснениям;
• полное спонтанное прекращение приступов. В подавляющем большинстве случаев отмечается у больных с криптогенными инфантильными спазмами.
Семиология эпилептических приступов при синдроме Веста.
Как уже говорилось, основным и единственным типом припадков при синдроме Веста являются инфантильные спазмы. Это один из кардинальных отличительных признаков заболевания от других ранних эпилептических энцефалопатий, в частности ранней миоклонической энцефалопатии. Спазмы представляют собой массивные генерализованные миоклонические или тонические сокращения аксиальной и конечностной мускулатуры.
Различают симметричные и асимметричные спазмы. При последних кинематика спазма имеет принципиально другой характер; наряду с типичными компонентами миоклонического или тонического инфантильного спазма отмечаются явные проявления парциального приступа, наиболее часто мимикрирующего асимметричный шейно-тонический рефлекс.
Типичный флексорно-экстензорный инфантильный спазм состоит из короткой флексии и следующей за ней форсированной экстензии указанных мышечных групп. Наиболее часто наблюдается смешанный характер задействования мышц при максимальной флексии аксиальных мышечных групп отмечаются относительная экстензия и абдукция рук. Очень часто приcтуп сочетается с различными приступными феноменами в мимической мускулатуре (по типу непроизвольных улыбок и гримас), вокализмами и разнообразными пароксизмальными вегетативными изменениями — покраснением лица, обильным потоотделением, тахикардией и т.д. Может отмечаться тоническая девиация глазных яблок вверх или в сторону, особенно часто в случаях асимметричных спазмов. Одним из патогномоничных симптомов в дифференциальной диагностике инфантильного спазма является наличие громкого, «страдальческого» постприступного плача.
Очень часто инфантильные спазмы при синдроме Веста ассоциированы типичные серии. Как правило, в серию может входить до 10—15 спазмой, дующих практически без перерыва один за другим во временном промежутке, не превышающем 10—12 мин. Эти серии получили очень удачное название — «пучки». Серийные спазмы отмечаются в 80 % случаев криптогенных инфантильных спазмов.
Продолжительность изолированного инфантильного спазма достаточно вариабельна и могут длиться от 0,5 до 2 сек.
Электроэнцефалографические аспекты синдрома Веста.
Лекция "1 Восточные славяне в VIII-IX веках" также может быть Вам полезна.
Основным (но не единственным) ЭЭГ-паттерном синдрома Веста является гипсаритмия. Гипсаритмия — это хаос и анархия на ЭЭГ.
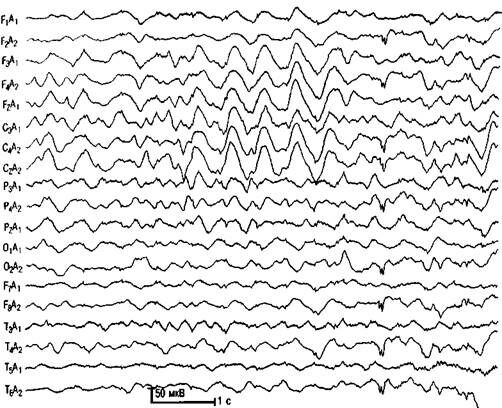
Типичная гипсаритмия - это выраженное диффузное замедление активности, наличие диффузной быстрой активности; асимметрия фоновой записи.
При трансформации синдром Веста в синдром Леннокса—Гасто типичная функциональная гипсаритмия постепенно и неуклонно замещается модифицированной с преобладаем межполушарной синхронизации или генерализованной медленноволновой активности.
X
Рис. 3.10. ЭЭГ ребенка 5 мес с инфантильными спазмами. Модифицированная гипсаритмия.


















