


Кинетика контактно-каталитических процессов превращения природных энергоносителей
16. Кинетика контактно-каталитических процессов превращения природных энергоносителей
Катализом называется изменение скорости химических реакций или возбуждение их в присутствии веществ — катализаторов, которые участвуют в реакции, вступая в промежуточное химическое взаимодействие с реагентами, но восстанавливают свой химический состав при окончании каталитического акта.
Обычно катализатор многократно вступает в такое взаимодействие, изменяя скорость химической реакции в течение длительного времени и образуя продукты реакции, вес которых может превосходить вес самого катализатора в тысячи и даже миллионы раз. Однако катализатор не может служить беспредельно: в одних промышленных процессах его используют непрерывно в течение нескольких лет, а в других — лишь несколько минут. Катализ может нарушиться в результате изменения состава и структуры катализатора вследствие побочных химических реакций или из-за механических и температурных воздействий. При возбуждении разветвленных цепных реакций, в частности, реакций, приводящих к взрыву, в принципе возможно и однократное участие катализатора в химической реакции.
Катализ называют положительным, если скорость реакции увеличивается. Отрицательный катализ означает уменьшение скорости химического превращения вследствие действия катализатора; он связан, как правило, с замедленным превращением в продукт промежуточного химического соединения.
Положительный катализ происходит тогда, когда скорость образования промежуточных соединений катализатора с реагентами и дальнейшего превращения их в продукт больше, чем скорость получения продукта некаталитическим путем. Практически катализаторы производят только для положительного катализа. Свободная энергия катализатора до акта катализа и после него неизменна. Поэтому в обратимых реакциях катализатор ускоряет достижение равновесия, но не смещает его.
Ускоряющее действие катализаторов весьма специфично и сильно отличается по эффективности и механизму воздействия от влияния других параметров процесса. Как известно, скорость технологического процесса можно повышать изменением температуры, давления, концентрации реагентов, применением перемешивания реагирующих масс и катализаторов.
Скорость процесса можно выражать изменением количества продукта, концентрации его и степени превращения основного исходного вещества во времени.
Повышением концентрации реагентов и давления увеличивают движущую силу процесса. Перемешивание приводит к увеличению скорости реакции только в тех случаях, в которых медленно происходит диффузия реагентов в зону реакции. Температура — наиболее универсальное средство интенсификации технологических процессов, повышение ее ускоряет химические реакции и в меньшей степени диффузию. Однако повышение температуры ограничено термостойкостью материалов и приводит к уменьшению движущей силы процесса в обратимых экзотермических процессах. Таким образом, интенсифицирующее действие всех параметров технологического режима ограничено за исключением действия катализаторов.
Сущность ускоряющего действия катализаторов состоит в понижении энергии активации Е химической реакции в результате изменения реакционного пути с участием катализатора или вследствие осуществления реакции по цепному механизму при инициирующем действии катализатора. Однако в некоторых типах каталитических реакций одновременно с понижением энергии активации происходит уменьшение предэкспоненциалыюго коэффициента в уравнении Аррениуса
Рекомендуемые материалы
В активации играет роль как кинетическая, так и внутримолекулярная энергия. Если общая энергия молекул превосходит Е, необходимую для реакции, то молекулы возбуждаются. При этом повышается не только скорость движения молекул, но и энергия колебания составляющих молекулу частиц. Изменяется электронное состояние молекулы, так как электроны переходят на более высокие энергетические уровни.
Одновременно с понижением энергии активации во многих случаях происходит понижение порядка реакции, которое объясняется тем, что в присутствии катализатора реакция идет через несколько элементарных стадий, порядок которых может быть меньше порядка некаталитических реакций.
Так реакция синтеза (А + В → С) в присутствии катализатора может идти через следующие элементарные стадии:
А + [К] = А`[К]
А`[К] +В = АВ`[К]
АВ`[К] = АВ` + [К]
На рис. 39 показано изменение энергии реагирующей системы для указанной реакции. Если Е – энергия активации некаталитической реакции, Ев – энергия активации каталитической реакции, е1 и е2 энергии активации промежуточных стадий, то Ев < Е – катализ положительный.
Каталитический процесс представляет собой совокупность каталитических реакций на поверхности катализатора с процессом подвода реагентов в зону реакции и отвода продуктов реакции.
В общем случае катализ на твердых катализаторах складывается из следующих элементарных стадий:
1. Эффективная внешняя диффузия реагирующих веществ и ядра потока к поверхности зерен катализатора. При этом коэффициент эффективной диффузии слагается из коэффициентов нормальной (молекулярной) диффузии и турбулентной (конвективной) диффузии. Последний называют также коэффициентом перемешивания, так как он выражает конвективный перенос вещества, вызванный турбулентным движение потока в слое катализатора.
2. Эффективная внутренняя диффузия в порах зерна катализатора в зависимости от соотношения размеров пор и молекул газов может проходить по нормальному молекулярному механизму или в стесненном движении.
3. Активированная (химическая) адсорбция одного или нескольких реагирующих компонентов на поверхности катализатора с образованием поверхностного химического соединения.
4. Перегруппировка атомов с образованием поверхностных комплексов: продукт — катализатор.
5. Десорбция продукта катализа (регенерация активного центра катализатора).
6. Диффузия продукта в порах зерна катализатора.
7. Диффузия продукта от поверхности зерна.
Каждая из стадий каталитического процесса должна быть охарактеризована энергией активации значительно меньшей, чем энергия активации гомогенной реакции, в противном случае протекание процесса каталитическим путем может оказаться энергетически невыгодным.
Общая скорость гетерогенного каталитического процесса отделяется относительными скоростями отдельных стадий и может лимитироваться наиболее медленной из них. Иногда скорость всего процесса определяют химические превращения на поверхности катализатора, а иногда диффузионные переносы веществ. Говоря о стадии, лимитирующей процесс, мы предполагаем, что остальные стадии протекают настолько быстро, что в каждой из них практически достигается равновесие; следовательно, полное изменение свободной энергии в них должно быть близко к нулю. Скорости отдельных стадий определяются параметрами технологического режима.
По механизму процесса в целом, включая собственно каталитическую реакцию и диффузионные стадии переноса вещества, различают процессы, проходящие в кинетической, внешнедиффузионной и внутридиффузионной областях.
В кинетической области константа скорости реакции не зависит от диффузии реагирующих веществ.
В кинетической области протекают главным образом процесс; на малоактивных катализаторах мелкого зернения с крупным порами при турбулентном течении потока реагентов, а также при низких температурах, близких к температуре зажигания катализатора. Однако для реакций в жидкостях переход в кинетическую область сопровождается понижением вязкости, а известно, что вязкость уменьшается с ростом температуры. С повышением температуры уменьшается также степень ассоциации, сольватации, гидратации молекул реагентов в растворах, что приводит к росту коэффициентов диффузии и соответственно к переходу из диффузионной области в кинетическую.
В последние годы создан процесс миллисекундного каталитического крекинга, осуществляемого на высокоактивном катализаторе в течение очень короткой продолжительности контакта паров сырья с катализатором. Процесс совершается в кинетической области.
Во внешнедиффузионной области протекают, прежде всего, процессы на высокоактивных катализаторах, обеспечивающих быструю реакцию и достаточный выход продукта за время контакт реагентов с катализаторами, измеряемое долями секунды. В этом случае нецелесообразно применять пористые зерна с высокоразвитой внутренней поверхностью катализатора, а нужно стремиться развить наружную поверхность катализатора.
Наиболее эффективным средством ускорения процессов, протекающих в области внешней диффузии, является перемешивать реагентов, которое в реакторе данной конструкции достигаете увеличением линейной скорости потока реагентов Сильная турбулизация потока приводит к переходу из внешне- во внутридиффузионную (при крупнозернистых мелкопористых катализаторах или же в кинетическую области.
Во внутридиффузионной области, т. е. когда общая скорости процесса лимитируется диффузией реагентов в порах зерна катализатора, существует несколько путей ускорения процесса. Можно уменьшать размеры зерен катализатора и соответственно путь молекул до середины зерна; это возможно, если одновременно переходят от фильтрующего слоя катализатора к кипящему. Можно изготовить для неподвижного слоя крупнопористые катализаторы, не уменьшая размеров зерен во избежание роста гидравлического сопротивления, но при этом неизбежно уменьшится внутренняя поверхность и соответственно понизится интенсивность работы катализатора по сравнению с мелкозернистым тонкопористым. Можно применять кольцеобразную контактную массу с небольшой толщиной стенок. Наконец, можно готовить бидисперсные или полидисперсные (мультидисперсные) катализаторы, в которых крупные поры являются транспортными путями к высокоразвитой поверхности, создаваемой тонкими порами малой длины (глубины).
Во всех этих случаях стремятся настолько уменьшить глубину проникновения реагентов в пору (и продуктов из поры), чтобы ликвидировать внутридиффузионное торможение и перейти в кинетическую область, когда скорость процесса определяется только скоростью собственно химических актов катализа, т. е. адсорбция реагентов активными центрами катализаторов, образования продукта и его десорбции.
Обычно лимитирующей стадией каталитического крекинга является собственно химическая реакция на поверхности (кинетическая область протекания реакции). В некоторых случаях для цеолитсодержащих катализаторов при неудовлетворительной пористой структуре матрицы скорость процесса лимитируется диффузией реагентов в порах (внутридиффузионная область протекания реакции).
Кроме пористой структуры матрицы диффузионное торможение могут оказывать также поры цеолитного компонента.
Крекинг индивидуальных углеводородов протекает в кинетической области, если они не содержат более двух циклов в молекуле и число атомов углерода в алкильной цепи не превышает 15 – 16. Соответственно для высококипящих нефтяных фракций превращение одной части компонентов будет протекать в кинетической области, а другой части – во внутридиффузионной или переходной части.
"Структура конфликта и его динамика" - тут тоже много полезного для Вас.
В связи с этим была разработана методика расчета процесса каталитического крекинга с учетом дезактивации цеолитсодержащего катализатора. Эти расчеты показали, что константы скорости крекинга тяжелого сырья выше, чем легкого из-за снижения стабильности нафтенов и парафинов с ростом их молекулярной массы. Селективность образования бензина и газа + кокса из нафтенов и парафинов приблизительно одинакова, и отношение бензин : (газ + кокс) составляет 5,7 – 5,8.
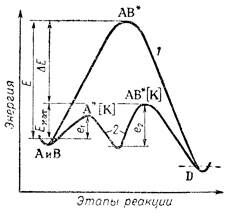
Рис. 39. Изменение энергии реагирующей системы при некаталитической (кривая I) и каталитической реакции (кривая II)
Нафтеновые углеводороды по сравнению с парафиновыми проявляют в 2,8 раза бóльшую реакционную способность. Алкилароматические углеводороды с наибольшей скоростью образуют газ и кокс, что обусловлено легкостью отщепления фрагментов С3 и С4 из алкильных групп и высокой тенденцией полициклических ароматических углеводородов к коксообразованию. Ароматические углеводороды без боковых цепей обнаруживают сильную склонность к коксообразованию, которая растет с увеличением их молекулярной массы.
Выход бензина проходит через максимум, величина которого падает в ряду циркулирующий газойль < ароматические углеводороды < парафины < нафтены. Скорость крекинга бензина мала, и отношение скоростей образования и распада составляет 14,3, что указывает на высокую селективность процесса. Предложенное кинетическое описание позволяет также рассчитать групповой состав газойлей, что важно для решения вопроса об их использовании.
В кинетической области скорость превращения исходного сырья определяется его концентрацией на поверхности в первой степени.













