
Термодинамическая вероятность различных направлений сложных реакций в процессах нефтепереработки
14. Термодинамическая вероятность различных направлений сложных реакций в процессах нефтепереработки
В соответствии с уравнением Гиббса (2.1) термодинамическая вероятность протекания химической реакции определяется знаком и величиной изменения свободной энергии Гиббса (изобарно-изотермического потенциала, свободной энтальпии). Изменения энергии Гиббса связано с константой равновесия реакции следующей формулой:
ln Kp =  ,
,
где Кр – константа равновесия, Кр = К1/К2, (К1 и К2 – константы скорости прямой и обратной реакций);  - изменение энергии Гиббса; R – газовая постоянная; Т – температура, К.
- изменение энергии Гиббса; R – газовая постоянная; Т – температура, К.
Если К1 > К2 (т.е. реакция идет в сторону образования продуктов), то Kp > 1 и ln Kp > 0, т.е.  < 0.
< 0.
Из уравнения (2.1) следует, что отрицательное значение (при низких температурах Т и давления Р) является условием самопроизвольного протекания химической реакции. Причем, чем больше абсолютное значение отрицательной величины , тем выше вероятность этой реакции.
Известно, что значение возрастает с увеличение молекулярной массы углеводородов (кроме ацетилена) и температуры. Следовательно высокомолекулярные углеводороды, имеющие большой потенциал образования , термически менее стабильны и более склонны к реакциям разложения, особенно при высоких температурах.
Для оценки термодинамической вероятности той или иной реакции применяют величину изменения свободной энергии, в результате реакции.
Свободной энергией называется та часть внутренней энергии системы, которая может быть превращена в работу. Реакции могут быть обратимыми и необратимыми. К обратимым реакциям, которые в зависимости от условий идут в одну или обратную сторону относят:
Рекомендуемые материалы
1. Образование простейших углеводородов из элементов и разложение углеводородов;
2. Гидрирование олефинов ↔ дегидрирование парафинов;
3. Гидрирование ароматики ↔ дегидрогенизация шестичленных нафтенов;
4. Конденсация ароматических углеводородов;
5. Изомеризация.
Многие реакции: крекинг, коксование, полимеризация являются необратимыми.
Для обратимых реакций любому значению внешних условий (температура и давление) отвечает некоторое состояние равновесной системы, характеризующееся определенным соотношением количеств исходных веществ и продуктов реакции. Это состояние равновесия оценивается константой равновесия.
Термодинамическая вероятность любой (в том числе и необратимой) реакции определяется знаком изменения величины свободной энергии реакции
 G (если известно значение G для любой изотермической реакции и если это значение оказывается положительным, в указанном направлении реакция термодинамически невозможна. Если же значение отрицательно, то процесс может происходить, и в действительности происходит, хотя бы даже с неизмеримо малой скоростью.
G (если известно значение G для любой изотермической реакции и если это значение оказывается положительным, в указанном направлении реакция термодинамически невозможна. Если же значение отрицательно, то процесс может происходить, и в действительности происходит, хотя бы даже с неизмеримо малой скоростью.
Взяв значения из таблиц значения свободных энергий образования из элементов начальных и конечных веществ G при каких либо температурах и по разнице определив для этих температур изменение свободной энергии реакции G , получаем коэффициенты А и В в уравнении
GТ = А + ВТ,
Т =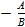
 G = 0, то это температурная граница термодинамической вероятности реакции: нижняя, если ∆G увеличивается с увеличением Т, и верхняя, если ∆G увеличивается с повышением температуры. Для обратимых реакций ре5акция идет и при температурах за пределами термодинамической вероятности, но с глубиной меньшей, чем у противоположной реакции. Впрочем, для обратимых реакций выходы можно изменить, меняя концентрации реагирующих веществ.
G = 0, то это температурная граница термодинамической вероятности реакции: нижняя, если ∆G увеличивается с увеличением Т, и верхняя, если ∆G увеличивается с повышением температуры. Для обратимых реакций ре5акция идет и при температурах за пределами термодинамической вероятности, но с глубиной меньшей, чем у противоположной реакции. Впрочем, для обратимых реакций выходы можно изменить, меняя концентрации реагирующих веществ.
Термодинамическая вероятность протекания химической реакции определяется знаком и величиной изменения свободной энергии Гиббса (изобарно-изотермического потенциала, свободной энтальпии). Изменение энергии Гиббса связано с константой равновесия реакции следующей формулой:
ln Кp = - ∆G/(RT),
где Кр — константа равновесия, Кр — K1/K2y (К1 и К2 — константы скорости прямой и обратной реакций); ∆ G — изменение энергии Гиббса; R— газовая постоянная; Т— температура, К.
Если К1 > K2 (т.е. реакция идет в сторону образования продуктов), то Кр > 1 и lпКр > 0, т.е. ∆G < 0.
Из уравнения следует, что отрицательное значение ∆G (при низких значениях температуры Т и давления Р) является условием самопроизвольного протекания химической реакции. Причем, чем больше абсолютное значение отрицательной величины ∆G, тем выше вероятность этой реакции.
Известно, что значение ∆G возрастает с увеличением молекулярной массы углеводородов (кроме ацетилена) и температуры. Следовательно, высокомолекулярные углеводороды, имеющие больший потенциал энергии образования ∆G, термически менее стабильны и более склонны к реакциям разложения, особенно при высоких температурах.
Промышленные термические процессы проводятся, как правило, под давлениями и сопровождаются гомогенными или гетерогенными реакциями.
В принципе, любые термические процессы нефтепереработки сопровождаются как эндотермическими реакциями дегидрирования и разложения углеводородов, так и экзотермическими реакциями синтеза, полимеризации, конденсации и т. п. Эти типы реакций различаются не только по знаку тепловых эффектов, но и по температурной зависимости значений свободной энергии Гиббса. Для эндотермических реакций разложения углеводородов значения ∆G уменьшаются с ростом температуры, а для экзотермических — увеличиваются, т. е. реакции разложения — термодинамически высокотемпературные, а синтеза — термодинамически низкотемпературные. Аналогичный вывод вытекает и из принципа Ле-Шателье — Брауна: повышение температуры способствует протеканию эндотермических реакций в сторону образования продуктов, а экзотермических — в обратную сторону.
В интервале температур 300—1200 °С, в котором осуществляется большинство промышленных процессов нефтепереработки, свободная энтальпия линейно зависит от температуры:
- ∆G = а + bT
В этом уравнении значение коэффициента b увеличивается с ростом теплового эффекта реакции (для эндотермических реакций b > 0, а для экзотермических b < 0). В реакциях с небольшим тепловым эффектом (например, изомеризации или гидрокрекинга) ∆G мало зависит от температуры. В реакциях же со значительным тепловым эффектом (выделение или поглощение) эта зависимость заметно значительнее.
Существенное влияние на величину константы скоростей реакций оказывает, в соответствии с принципом Ле-Шателье — Брауна, давление. Его рост способствует протеканию реакций с уменьшением объема (в основном реакции синтеза). Низкие же давления ускоряют реакции разложения.
Прогнозирование вероятности образования того или иного продукта разложения при осуществлении термических процессов также может базироваться на основе термодинамических данных, в частности, на значениях энергии связи между атомами в молекулах. Так, анализ данных по свободной энтальпии образования позволяет сделать следующие выводы о направлении разложения углеводородов.
1. В молекулах алканов энергия разрыва связи между крайним атомом углерода и водородом наибольшая в метане (431 кДж/моль), и она снижается по мере увеличения числа углеродных атомов до 4 и затем становится постоянной (на уровне 394 кДж/моль).
2. В нормальных алканах энергия разрыва связи между атомами водорода и находящегося внутри цепи углерода постепенно уменьшается в направлении к середине цепи (до 360 кДж/моль).
3. Энергия отрыва атома водорода от вторичного, и особенно от третичного атома углерода несколько меньше, чем от первичного.
4. В молекуле алкенов энергия отрыва атома водорода от углеродного атома с двойной связью значительно больше, а от атома углерода, находящегося в сопряжении с двойной связью, — значительно ниже, чем энергия С—Н-связи в алканах.
5. В нафтеновых кольцах прочность связи С—Н такая же, как в связях вторичного атома углерода с водородом в молекулах алканов. 6.
6. В молекулах бензола и алкилароматических углеводородов энергия связи между атомом углерода в кольце и водородом сопоставима с прочностью С—Н-связи в метане, а энергия отрыва водорода от углерода, сопряженного с ароматическим кольцом, значительно ниже, чем энергия С—Н-связи в алканах.
7. Энергия разрыва углерод-углеродной связи в молекулах всех классов углеводородов всегда ниже энергии С—Н-связи (примерно на 50 кДж/моль).
8. В молекулах алканов длина, строение цепи и местоположение разрываемой связи оказывают влияние на энергию разрыва углерод-углеродной связи качественно, аналогично влиянию их на прочность С—Н-связи. Так, связь между крайними углеродными атомами ослабляется по мере увеличения числа углеродных атомов (от 360 для этана до 335 кДж/моль для пентана и выше), а связь между внутренними углеродными атомами — по мере приближения к середине цепи (до 310 кДж/моль). Например, энергия разрыва связи С—С в молекуле н-октана в зависимости от ее местоположения изменяется следующим образом: 335; 322; 314; 310; 314; 322; 335 кДж/моль.
9. Связи между первичными атомами углерода всегда прочнее, чем С—С-связи в комбинациях с первичным, вторичным (Свт) и третичным (Стр) атомами углерода. Энергия разрыва углерод-углеродной связи (Dc-c) уменьшается в следующей последовательности:
DC-C > DC-Cвт > DC-Cтр > DCвт-Cвт > DCвт-Cтр > DCтр-Cтр.
10. В алкенах углерод-углеродные двойные связи значительно прочнее (но менее чем в 2 раза), чем С—С-связи в алканах. Так, энергия разрыва С = С-связи в этилене составляет 500 кДж/моль. Однако С—С-связи, сопряженные с двойной (т. е. находящиеся к ней в b-положении), значительно слабее С—С-связи в алканах.
11.Энергия разрыва углерод-углеродной связи в кольце циклопентана (293 кДж/моль) и циклогексана (310 кДж/моль) несколько меньше С—С-связи в середине цепи нормального гексана (318 кДж/ моль).
12. В алкилароматических углеводородах углерод-углеродная связь, сопряженная с ароматическим кольцом (С—Сар), менее прочна, чем связь С—С в алканах. Сопряжение с ароматическим кольцом снижает прочность углерод-углеродной связи приблизительно в такой же степени, как и сопряжение с двойной связью. Сопряжение с несколькими бензольными кольцами снижает прочность С-С-связи еще больше.
Рекомендуем посмотреть лекцию "Типология кочевого хозяйства".
13.Энергия разрыва (диссоциации) атомов водорода в молекуле водорода несколько выше С—Н-связи в наиболее термостойком метане и составляет 435 кДж/моль.
14. По прочности связь С—S в меркаптанах и связь S—S в дисульфидах сопоставима со связью С—С в алканах.
Очевидно, что при термолизе углеводородного сырья будут разрываться в первую очередь наиболее слабые связи и образовываться продукты преимущественно с меньшей свободной энергией образования. Таким образом, термодинамический анализ позволяет прогнозировать компонентный состав и подсчитать равновесные концентрации компонентов в продуктах реакций в зависимости от условий проведения термических, а также каталитических процессов.
Схемы (I—IV) химических превращений углеводородов при термолизе представлены на рис. 38.
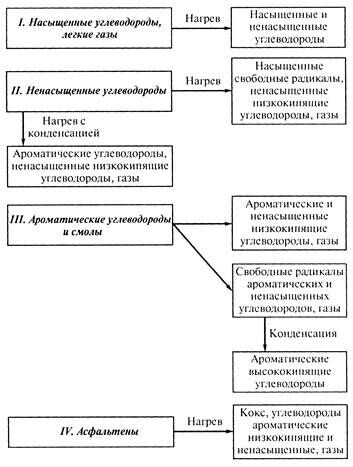
Рис. 38. Схемы превращений углеводородов при термолизе сырья


















